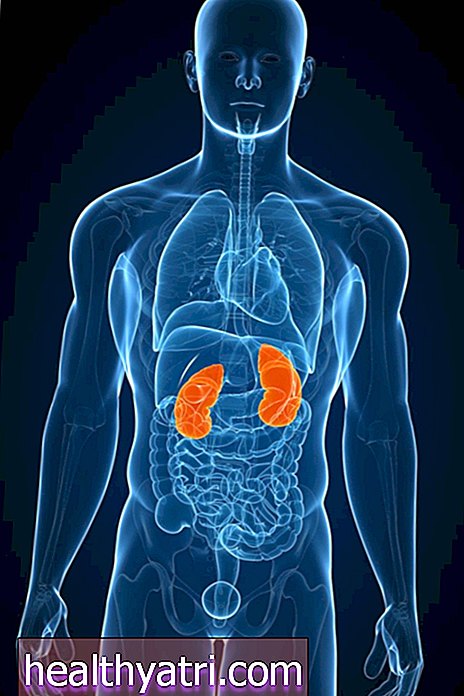
La enfermedad renal poliquística, o PKD, es una forma genética específica de enfermedad renal. Como sugiere el término, "poli" -quístico se refiere a la presencia de múltiples quistes (sacos cerrados y vacíos, a veces llenos de líquido) en el riñón. Los quistes renales en general no son un hallazgo infrecuente, pero un diagnóstico de quistes en el riñón no es necesariamente PKD.
La PKD, de hecho, es solo una de las múltiples razones por las que una persona puede desarrollar quistes en el riñón. Es la herencia genética específica y el curso de la PKD lo que la convierte en una entidad muy específica. No es una enfermedad benigna, y una gran parte de los pacientes podrían ver cómo sus riñones se deterioran hasta fallar, lo que requiere diálisis o un trasplante de riñón.
Mohammed Haneefa Nizamudeen / Getty ImagesOtros tipos de quistes
El otro tipo de quistes renales (que no son quistes relacionados con la PKD) incluyen:
- Quistes simples benignos, que suelen ser un resultado benigno del proceso de envejecimiento. Casi el doce por ciento de las personas de 50 a 70 años y el 22,1 por ciento de todas las personas mayores de 70 años tendrán al menos un quiste en el riñón.
- Maligno (cuando los quistes pueden ser representativos de cáncer en los riñones, a veces llamados quistes complejos).
- Adquirido, como en pacientes con enfermedad renal crónica (ERC).
Por lo tanto, una vez que se observan quistes en un riñón, el siguiente paso es diferenciar si se trata de un hallazgo benigno relacionado con la edad, PKD u otra cosa.
Genética
La PKD es un trastorno genético relativamente común, que afecta a casi 1 de cada 500 personas y sigue siendo una de las principales causas de insuficiencia renal.La enfermedad generalmente se hereda de uno de los padres (90 por ciento de los casos) o, más raramente, se desarrolla "de-novo" (llamada mutación espontánea).
Comprender la genética de la PKD es esencial para comprender los síntomas y el curso de la enfermedad. El modo de herencia de padres a hijos diferencia entre los dos tipos de PKD.
La PKD autosómica dominante (AD-PKD) es la forma hereditaria más común y el 90 por ciento de los casos de PKD son de este tipo. Los síntomas generalmente se desarrollan más tarde en la vida alrededor de los 30 a 40 años, aunque no se desconoce la presentación en la infancia.
Los genes anormales podrían ser los denominados genes PKD1, PKD2 o PKD3. Cuál de estos genes tiene la mutación y qué tipo de mutación podría ser tiene un efecto enorme en el resultado esperado de la PKD. Por ejemplo, el gen PKD1, que se encuentra en el cromosoma 16, es el sitio de mutación más común observado en el 85 por ciento de los casos de PQRAD. Los defectos en el gen (como es el caso de otras mutaciones también) conducen a un mayor crecimiento de células epiteliales en el riñón y la consiguiente formación de quistes.
La PKD autosómica recesiva (AR-PKD) es mucho más rara y podría comenzar temprano, incluso mientras el bebé se desarrolla durante el embarazo. Una de las razones por las que este tipo de PKD es poco común es porque los pacientes afectados generalmente no vivirán lo suficiente para procrear y transmitir la mutación a sus hijos.
Nuevamente, para resumir, el 90 por ciento de los casos de PKD se heredan, y de los tipos heredados, el 90 por ciento son autosómicos dominantes. Por lo tanto, los pacientes con PKD suelen tener PKD autosómica dominante (AD-PKD).
Ubicación de la gravedad y la mutación
El sitio de la mutación tendrá un impacto en el curso de la enfermedad. Con la mutación PKD2, los quistes se desarrollan mucho más tarde y, por lo general, la insuficiencia renal no ocurre hasta mediados de los 70. Compare esto con las mutaciones del gen PKD1, donde los pacientes pueden desarrollar insuficiencia renal a mediados de los 50.
Los pacientes con mutaciones de PKD2 a menudo ni siquiera conocerán ningún antecedente familiar de PKD. En este caso, siempre es posible que el antepasado portador de la mutación muriera antes de que la enfermedad fuera lo suficientemente grave como para causar síntomas o requerir diálisis.
Síntomas
Se pueden observar una variedad de síntomas en la PKD. Los ejemplos comunes incluyen:
- Dolor en el flanco debido al agrandamiento de los riñones.
- Infecciones del tracto urinario
- Cálculos renales (debido al flujo lento de orina en los quistes)
- Los quistes también pueden estar presentes en otros órganos como el hígado y el páncreas.
- Los pacientes tienden a tener presión arterial alta debido al papel de los riñones en la regulación de la presión arterial.
Diagnóstico
Aunque las mutaciones de la PKD suelen estar presentes al nacer, es posible que los quistes renales no sean evidentes en ese momento. Estos quistes se convierten en sacos apreciables llenos de líquido durante las primeras dos décadas, momento en el que pueden comenzar a causar síntomas o signos cuando alguien llega a los 30 años. Sin embargo, el avance de la enfermedad renal hasta el punto de fallar podría llevar décadas. a partir de entonces.
La mayoría de las personas que conocen antecedentes familiares de PKD tienen un umbral bajo para ser diagnosticadas con PKD, ya que tanto los pacientes como los médicos conocen bien la fuerte naturaleza familiar de la enfermedad. En los casos en los que los antecedentes familiares pueden no conocerse o son aparentemente "normales", el diagnóstico es más difícil y requiere la evaluación de un nefrólogo. En este caso, el padre afectado podría haber muerto antes de que la enfermedad tuviera la oportunidad de progresar a una enfermedad renal en etapa terminal. Finalmente, si se trata de un caso de "mutación espontánea", es posible que no haya PKD presente en ninguno de los padres.
El diagnóstico inicial de PKD se realiza mediante estudios de imágenes como ultrasonido o tomografía computarizada. Sin embargo, el hecho de que alguien tenga múltiples quistes en los riñones no significa necesariamente que tenga PKD. Podría ser simplemente un caso de uno-demasiados quistes simples u otras posibilidades como la enfermedad renal quística medular (no lo mismo que la PKD).
Cuando el diagnóstico está en duda, las pruebas genéticas pueden confirmar o refutar el diagnóstico. Sin embargo, las pruebas genéticas tienden a ser costosas y, por lo tanto, se usan principalmente cuando el diagnóstico es ambiguo.
Curso de la enfermedad
¿Cuánto tiempo tardan las personas con PKD en desarrollar insuficiencia renal? Esta es quizás la pregunta número uno que tendrán las personas recién diagnosticadas con PKD. En el peor de los casos, donde los pacientes avanzan hasta la insuficiencia renal completa, que requieren diálisis o trasplante, la función renal (TFG) podría disminuir en alrededor de 5 puntos por año. Por lo tanto, alguien que comienza con una TFG de 50 podría llegar a una TFG de cinco en aproximadamente nueve años, momento en el que ciertamente se podría requerir diálisis o trasplante.
Tenga en cuenta que no todos los pacientes con PKD necesariamente disminuirán hasta la insuficiencia renal completa. Lo que aún debe enfatizarse es que no todas las personas con PKD necesariamente progresarán hasta el punto en que necesiten diálisis. Los pacientes con mutación del gen PKD2 obviamente tienen más posibilidades de evitar una insuficiencia renal completa. Esta es la razón por la que, en conjunto, menos de la mitad de los casos de PKD se diagnosticarán durante la vida del paciente, ya que la enfermedad podría ser clínicamente silenciosa.