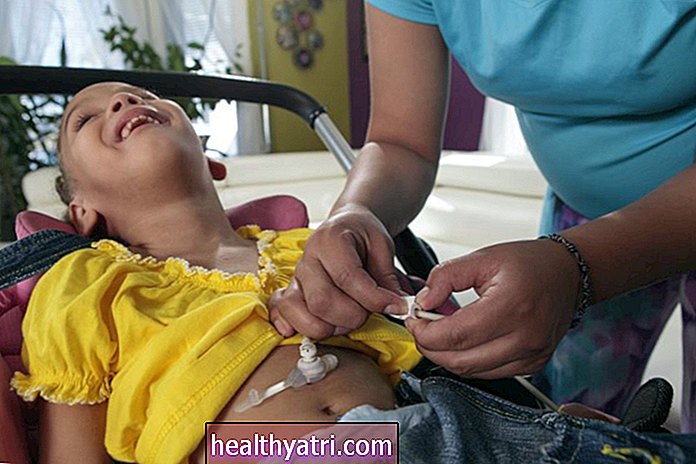
La fibrosis quística es un trastorno hereditario y potencialmente mortal que afecta aproximadamente a 30.000 estadounidenses y hasta 100.000 personas en todo el mundo. Es causada por un defecto genético en el gen del receptor transmembrana de la fibrosis quística (CFTR), que crea la proteína involucrada en la producción de sudor, fluidos digestivos y moco. Si hay un defecto en esta proteína, puede provocar la acumulación anormal de moco en los pulmones, el bloqueo de las enzimas digestivas en los intestinos y otros síntomas y complicaciones graves.
Hay más de 2000 mutaciones CFTR conocidas. Para que tenga fibrosis quística, debe haber heredado dos copias de la mutación CFTR, una de cada padre. Si bien los avances en el diagnóstico y el tratamiento han aumentado la esperanza de vida de las personas que viven con la enfermedad, todavía no existe cura.
Ilustración de Verywell
Genética
La fibrosis quística (FQ) es un trastorno autosómico recesivo. Este es el tipo de enfermedad que solo puede heredar si ambos padres aportan una sola copia de un gen recesivo (en este caso, la mutación CFTR).
Por definición, un gen recesivo es aquel que puede estar enmascarado por un gen dominante. Un ejemplo son los ojos azules, que es un rasgo recesivo, y los ojos marrones, que es un rasgo dominante. Si solo hereda un gen recesivo, no exhibiría el rasgo recesivo, sino que sería portador del gen.
Con respecto a la FQ, puede heredar la enfermedad si cada uno de sus padres tiene una mutación de CFTR o la propia FQ. Por otro lado, si uno de sus padres es portador y el otro tiene FQ, usted tiene una probabilidad del 50/50 de tener FQ o de ser portador. Desafortunadamente, no hay nada que un padre pueda hacer para influir en las probabilidades de herencia de una forma u otra.
Riesgo de heredar fibrosis quística
Si ambos padres son portadores de la mutación del gen CFTR, tendría:
- 25 por ciento de probabilidad de heredar dos copias y tener FQ
- 50 por ciento de posibilidades de heredar una copia y ser portador
- 25 por ciento de probabilidad de no heredar mutaciones y no verse afectado
Raza y etnia
La fibrosis quística es más común entre las personas de ascendencia europea del norte y afecta a uno de cada 3.000 recién nacidos. Es menos común en personas de ascendencia africana o asiática y afecta a las mujeres solo un poco más que a los hombres.
En los Estados Unidos, la tasa de FQ varía según la población racial. Se estima que la cantidad de estadounidenses que portan una mutación de CFTR es de alrededor de:
- 1 de cada 29 estadounidenses de raza blanca (1 de cada 3500 de riesgo)
- 1 de cada 46 hispanoamericanos (riesgo de 1 de cada 10,000)
- 1 de cada 65 afroamericanos (riesgo de 1 de cada 20.000)
- 1 de cada 90 asiático-americanos (1 de cada 100.000 de riesgo)
La tasa real de bebés que nacen con FQ es de alrededor de:
- 1 de cada 3500 estadounidenses de raza blanca
- 1 de cada 10,000 hispanoamericanos
- 1 de cada 20.000 afroamericanos
- 1 de cada 100.000 estadounidenses de origen asiático
El país con mayor tasa de bebés nacidos con FQ es Irlanda, en el que se verá afectado uno de cada 1.353 nacimientos, según un estudio epidemiológico publicado en elRevista de investigación biomédica y biotecnológica.
Tipos de mutaciones CFTR
No todas las mutaciones de CFTR son iguales. Las mutaciones se dividen en seis clases según las características del defecto y su impacto en el organismo. Las clases 1, 2 y 3 darán como resultado los síntomas "clásicos" más graves de la FQ, mientras que las clases 4, 5 y 6 son más leves en comparación.
La función de la proteína CFTR es controlar el movimiento del agua y la sal dentro y fuera de las células. Al hacerlo, ayuda a regular la producción de moco, sudor, saliva, lágrimas y enzimas digestivas. Según cuán defectuosa sea la proteína, estos sistemas pueden funcionar mal de maneras a menudo graves.
Las clases de mutaciones de CFTR se pueden describir ampliamente de la siguiente manera:
- Clase 1: la mutación da como resultado la producción de pocas o ninguna CFTR.
- Clase 2: la mutación hace que CFTR se deforme y no sea funcional.
- Clase 3: la mutación causa un "defecto de activación" en el que CFTR bloquea el movimiento de agua y sal dentro y fuera de las células.
- Clase 4: la mutación causa un "defecto de conductancia" donde CFTR restringe el movimiento de la sal dentro y fuera de las células.
- Clase 5: la mutación disminuye la producción de proteína CFTR.
- Clase 6: la mutación da como resultado un CFTR funcional e inestable que necesita ser reemplazado constantemente.
Diferentes combinaciones de mutaciones pueden conducir a diferentes clases de enfermedades. Un ejemplo de ello es la mutación deltaF508 que se observa en alrededor del 70 por ciento de los casos. Si hereda el deltaF508 de ambos padres, tendría una característica CFTR no funcional de la enfermedad de clase 2. Otros genes, llamados genes modificadores, pueden degradar aún más la función de las proteínas y provocar un empeoramiento de los síntomas.
Fisiología
Para comprender cómo los defectos de CFTR causan fibrosis quística, debe observar más de cerca los sistemas que la proteína debe regular.
La proteína CFTR se conoce como proteína de canal. Es producido por el cuerpo con el único objetivo de mantener el equilibrio de agua y sal en las células. En circunstancias normales, si hay algo que afecte este equilibrio, CFTR moverá agua y sal dentro y fuera de la célula para mantener la estasis (equilibrio).
Con la fibrosis quística, la proteína CFTR funciona de manera anormal. En lugar de mover agua dentro y fuera de las células, el agua queda atrapada, lo que hace que el moco fuera de la célula se espese y se acumule.
Esta acumulación interferirá con el funcionamiento normal de los órganos de diferentes formas:
- En los pulmones, la acumulación de moco puede bloquear las vías respiratorias, provocando inflamación y aumentando el riesgo de infección, hipertensión pulmonar y daño tisular.
- En el tracto digestivo, la acumulación puede bloquear la secreción de enzimas digestivas del páncreas. Esto puede interferir con la absorción de nutrientes en los intestinos, provocando desnutrición y un crecimiento deficiente. También puede ocurrir pancreatitis crónica.
- En el hígado, la obstrucción de los conductos biliares puede interferir con la capacidad del hígado para eliminar las toxinas de la sangre, provocando cicatrices, cálculos biliares y cirrosis.
- En el sistema endocrino, el bloqueo de las células productoras de insulina en el páncreas, conocidas como islotes de Langerhans, puede conducir a un tipo de diabetes que tiene características tanto de diabetes tipo 1 como tipo 2. Esto se llama diabetes relacionada con la fibrosis quística (CFRD).
Guía de discusión del médico sobre fibrosis quística
Obtenga nuestra guía imprimible para su próxima cita con el médico para ayudarlo a hacer las preguntas correctas.

Envíelo a usted mismo oa un ser querido.
InscribirseEsta Guía de discusión para médicos se envió a {{form.email}}.
Hubo un error. Inténtalo de nuevo.
Enfermedad progresiva
El único factor de riesgo para la FQ es tener dos padres que portan genes CFTR anormales. Dicho esto, existen factores que pueden influir en la gravedad y progresión de la enfermedad.
El principal de ellos es el momento del diagnóstico y el tratamiento. El cribado neonatal se considera vital ya que permite el tratamiento inmediato de la enfermedad. Al hacerlo, podemos retrasar o prevenir el daño que puede ocurrir desde los dos primeros meses de vida.
Según un estudio publicado enOpiniones actuales en medicina pulmonar, los niños que reciben tratamiento después de que aparecen los síntomas de la FQ generalmente tendrán un deterioro significativo del flujo de aire y signos de lesión respiratoria a la edad de dos años. En comparación, los niños identificados y tratados al nacer tendrán, a la edad de dos años, una función pulmonar comparable a la de un niño de un año en el grupo de tratamiento retrasado.
El tratamiento temprano, junto con los avances en las terapias con medicamentos, significa que los niños diagnosticados con FQ en la actualidad pueden vivir hasta bien entrados los 40 y 50 años y, en gran medida, no se ven afectados por la enfermedad.
A pesar de los avances en el diagnóstico y el tratamiento, persisten desafíos.Al final, la fibrosis quística está influenciada tanto por cosas que podemos controlar como por cosas que no podemos.
Entre algunos de los factores de riesgo relacionados con peores resultados:
- El crecimiento deficiente es el factor más fuertemente asociado con la enfermedad pulmonar FQ grave, según una investigación de la Universidad de Wisconsin. Para ello, las personas con FQ necesitan consumir una gran cantidad de calorías para mantener el peso y el crecimiento, lo que a menudo es difícil si existe una afectación intestinal grave.
- El subtratamiento de antibióticos es otro factor de riesgo común. Debido al alto riesgo de infecciones bacterianas, las personas con FQ suelen recibir antibióticos profilácticos (preventivos) incluso si están sanos. El subtratamiento puede aumentar el riesgo de infección, mientras que el uso inconsistente de antibióticos a largo plazo puede generar resistencia, lo que limita las opciones de tratamiento en el futuro.
- Pseudomonas aeruginosaLa infección, una bacteria comúnmente adquirida por personas que han estado en el hospital durante más de una semana, se asocia con una progresión rápida de la enfermedad. Como tal, la hospitalización se considera un factor de riesgo independiente para la progresión de la FQ.
- El consumo de alcohol puede acelerar el daño hepático al tiempo que aumenta el riesgo de pancreatitis aguda o crónica, según una investigación publicada en la revista.Gastroenterología.
- El humo de segunda mano aumenta el riesgo de infecciones y complicaciones casi tanto como fumar. Según una investigación de la Escuela de Medicina de la Universidad John Hopkins, el humo de segunda mano se asocia con una disminución del 10 por ciento en la capacidad y función pulmonar. Si bien esto puede no parecer una pérdida grave, en última instancia significa que un joven de 17 años con FQ expuesto al humo de segunda mano tendría la misma función pulmonar que un joven de 24 años con FQ que no ha estado expuesto.

-overview.jpg)