La atrofia muscular espinal (AME) se puede diagnosticar con pruebas genéticas.Si usted o su hijo padecen la afección, puede llevar semanas, meses o incluso más tiempo confirmar que la AME es la causa de síntomas como debilidad muscular y dificultad para respirar. Si su equipo médico está preocupado por la AME, se puede solicitar una prueba genética. Varios estados examinan rutinariamente a los recién nacidos para detectar AME, y los defensores de la detección de AME en recién nacidos creen que la lista está creciendo.
Existen otras afecciones que pueden causar efectos similares a los de la AME, y es posible que usted o su hijo necesiten una evaluación médica que también incluya pruebas de diagnóstico para otras afecciones.
Imágenes de JohnnyGreigg / Getty
Autocomprobaciones / pruebas en el hogar
Las señales de advertencia de AME que debe buscar en el hogar difieren según la edad a la que la afección comienza a tener efectos clínicos. Si le preocupa que usted o su hijo puedan tener músculos débiles, hay varias cosas que puede hacer para verificar si necesita ver a un médico.
Bebés y niños pequeños
Es posible que los nuevos padres que no hayan tenido otros hijos antes no sepan qué esperar en términos de los movimientos de un bebé. Está perfectamente bien si no está seguro de si hay un problema; si está preocupado, es mejor pedir ayuda. El pediatra de su hijo podrá reconocer los primeros signos de AME.
Un niño que tiene AME tendría las siguientes características:
- Dificultad para comer: su bebé puede tener dificultad para tragar, succionar o mover la cabeza hacia un biberón o tetina.
- Movimientos musculares: es posible que su bebé no mueva espontáneamente los brazos y las piernas, estire el cuerpo, alcance objetos o gire la cabeza.
- Tono muscular bajo: los músculos de su bebé pueden parecer débiles y flácidos, y sus brazos o piernas pueden caer cuando no los está levantando. Por ejemplo, si levanta los brazos de su bebé durante un baño de esponja, sus brazos pueden caer cuando lo suelte. O si levanta las piernas de su bebé para cambiarle el pañal, es posible que se le caigan las piernas cuando lo suelte.
- Sentarse sin ayuda: los bebés que tienen AME de inicio muy temprano (tipo cero o tipo uno) no aprenderán a sentarse. Los bebés que tienen AME tipo dos pueden aprender a sentarse y luego perder esa capacidad.
- Dificultad para respirar: puede notar que su bebé está respirando poco profundo o esforzándose mucho para respirar.
Adolescentes y adultos
Los tipos tres y cuatro de AME de aparición tardía comienzan al final de la infancia, la adolescencia o la edad adulta. Puede notar problemas para subir escaleras o levantar objetos pesados o grandes. A veces, pueden ocurrir espasmos musculares.
El agotamiento puede ser el problema principal con estos tipos de AME de aparición tardía.
En general, los signos de AME en el hogar no son específicos: usted sabe que algo anda mal, pero no necesariamente lo que es. Si usted o su hijo experimentan debilidad muscular, espasmos, problemas respiratorios o agotamiento, asegúrese de consultar al médico lo antes posible.
Monitoreo en casa
Es posible que los bebés, los niños y los adultos que tienen AME necesiten que se controlen los niveles de oxígeno en casa con un dispositivo no invasivo. Este dispositivo, que se coloca en el dedo, puede aproximar los niveles de oxígeno en la sangre.
Su médico puede recomendar su uso todo el tiempo, o al dormir, para detectar una caída repentina en el nivel de oxígeno en sangre.
Laboratorios y pruebas
Una prueba genética, que se realiza en una muestra de sangre, es la prueba más definitiva de AME. Si usted o sus médicos están preocupados por la posibilidad de AME, probablemente se necesite una prueba genética.
Si tiene antecedentes familiares de AME, la prueba genética se puede realizar incluso antes de que comiencen los síntomas. De hecho, la detección de AME de todos los recién nacidos es estándar en varios estados, incluso para los bebés que no tienen antecedentes familiares de la afección.
Si su equipo médico descarta otras afecciones además de la AME, es posible que también deba realizarse otras pruebas de diagnóstico.
El diagnóstico oportuno se considera ventajoso porque se cree que los tratamientos utilizados para el manejo de la AME son más efectivos cuando se inician temprano. Además, las complicaciones como las emergencias respiratorias y las infecciones se pueden prevenir cuando la enfermedad se reconoce en una etapa temprana.
Análisis de sangre
Los análisis de sangre se utilizan para identificar la causa de la debilidad muscular en niños y adultos o para controlar la función respiratoria en la AME.
Creatina quinasa: si muestra signos de debilidad muscular o problemas respiratorios, es posible que le realicen análisis de sangre, como el nivel de creatina quinasa. Esta proteína puede elevarse cuando una persona tiene daño muscular, lo que puede ocurrir en algunas afecciones neuromusculares. Se espera que la creatina quinasa sea normal o casi normal en la AME.
Gasometría arterial: si la respiración se ha convertido en un problema, los niveles de oxígeno se pueden medir con precisión utilizando sangre extraída de una arteria. Esta prueba generalmente se realiza en un hospital o en un entorno de rehabilitación cuando una persona está recibiendo oxígeno o ayuda para respirar o tiene un riesgo inminente de tener problemas respiratorios.
Prueba genética
Esta prueba se realiza con un simple análisis de sangre no invasivo. Varias miopatías hereditarias (enfermedades musculares) y afecciones metabólicas pueden causar síntomas similares a los de la AME, y su médico también puede enviar pruebas genéticas para cualquier otra afección potencial que usted pueda tener.
La prueba genética puede identificar una mutación (alteración) en el gen SMN1, que se encuentra en el cromosoma 5. Si una persona tiene la mutación en ambas copias del cromosoma 5 (una del padre y otra de la madre), se espera que desarrollar los efectos físicos de la AME.
La prueba genética también identifica la cantidad de copias del gen SMN2, que también se encuentra en el cromosoma 5. Si una persona tiene pocas copias, se espera que los efectos de la AME comiencen temprano en la vida y sean severos. Si una persona tiene muchas copias (hasta ocho o 10), se espera que la afección comience más tarde en la vida y tenga efectos leves.
Una persona se considera portadora de AME si una de sus copias del cromosoma 5 tiene un gen SMN1 con la alteración genética. Un portador puede transmitir el gen a sus hijos si el niño también recibe otro gen SMN1 alterado del otro padre.
También hay algunos otros genes que pueden causar AME: eldineína citoplasmática 1 cadena pesada 1(DYNC1H1) en el cromosoma 14 o el gen de la enzima activadora de ubiquitina 1 (UBA1) en el cromosoma X. Una persona que hereda una copia defectuosa de cualquiera de estos genes desarrollará AME.
Imágenes, estudios eléctricos y biopsia
Las pruebas de imagen no son especialmente útiles en el diagnóstico de AME. Al igual que con algunas de las otras pruebas de diagnóstico, generalmente solo son necesarias si existe una preocupación sobre otros posibles diagnósticos.
A medida que avanza la afección, a menudo se necesitan pruebas por imágenes para evaluar complicaciones, como cambios en la columna ósea e infecciones.
Las pruebas por imágenes que se pueden utilizar en la evaluación y el tratamiento de la AME incluyen:
- Resonancia magnética cerebral: una resonancia magnética cerebral puede mostrar cambios anatómicos. Se espera que esta prueba sea normal en la AME, pero varias de las otras enfermedades que causan debilidad (como la adrenoleucodistrofia cerebral) están asociadas con cambios en una resonancia magnética cerebral.
- Radiografía de la columna vertebral: a menudo, se usa una radiografía de la columna vertebral para diagnosticar la escoliosis. Esto puede ir seguido de una resonancia magnética de la columna si se necesita una evaluación adicional.
- Resonancia magnética de la columna: no se esperaría que una resonancia magnética de la columna muestre cambios que ayuden en el diagnóstico de AME, pero puede mostrar cambios asociados con complicaciones de la AME, como la escoliosis.
- Radiografía de tórax: una radiografía de tórax suele ser útil para identificar la neumonía, que puede ocurrir debido a la debilidad de los músculos respiratorios de la AME.
Estudios Eléctricos
La electromiografía (EMG) y los estudios de velocidad de conducción nerviosa (NCV) son estudios eléctricos de diagnóstico que se utilizan a menudo en la evaluación de la debilidad muscular.
La NCV es una prueba no invasiva que utiliza descargas eléctricas colocadas en la piel para evaluar la función nerviosa motora y sensorial según la velocidad registrada del nervio. Un EMG implica la colocación de una aguja fina en el músculo para medir su función.
Ambas pruebas pueden ser un poco incómodas, especialmente para un niño pequeño. Sin embargo, debe estar seguro de que estas pruebas eléctricas son seguras, útiles y no causan ningún efecto secundario.
La EMG y la NCV pueden mostrar patrones diferentes dependiendo de si una persona tiene una enfermedad muscular, una enfermedad de los nervios periféricos o una enfermedad de las neuronas motoras. Una EMG o NCV pueden mostrar evidencia de un déficit de neuronas motoras en personas que tienen AME, aunque estas pruebas no siempre son anormales en la AME.
La EMG puede mostrar evidencia de denervación (pérdida de estimulación nerviosa de un músculo) y fasciculaciones (contracciones musculares diminutas), mientras que la NCV puede mostrar evidencia de función nerviosa motora más lenta. Se espera que las medidas de la función del nervio sensorial sean normales en la AME.
Biopsia
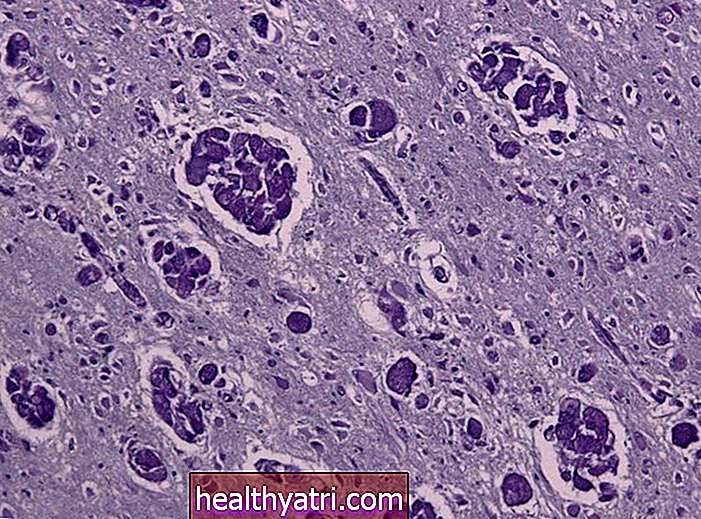
Las biopsias del nervio, el músculo o la médula espinal pueden mostrar anomalías en la AME, pero estas pruebas no suelen ser necesarias. La prueba genética para la AME no es invasiva y es confiable, mientras que una biopsia es un procedimiento invasivo cuyos resultados no siempre ayudan a verificar o descartar la AME.
Se esperaría que una biopsia de músculo mostrara signos de atrofia (encogimiento del músculo). Una biopsia de nervio puede ser normal o mostrar signos de degeneración nerviosa. Y una biopsia del cuerno anterior de la columna mostraría una atrofia severa de las células de las neuronas motoras.
Diagnóstico diferencial
Hay una serie de afecciones neuromusculares y metabólicas que pueden causar debilidad muscular y disminución del tono muscular. Las otras enfermedades consideradas en el diagnóstico diferencial de AME son diferentes para los niños que para los adultos porque algunas de estas enfermedades generalmente comienzan durante la infancia, mientras que otras comienzan durante la edad adulta.
Condiciones médicas que pueden tener características similares a las de la AME, que incluyen:
Miopatía (enfermedad de los músculos): existen muchos tipos de miopatía. La gravedad de la debilidad muscular varía según los diferentes tipos. Es posible que se necesiten pruebas de diagnóstico con análisis de sangre, estudios eléctricos y posiblemente una biopsia si se considera que la miopatía es una posible causa de sus síntomas.
Distrofia muscular: la distrofia muscular es un subconjunto de la miopatía; Hay nueve tipos de distrofia muscular, incluida la distrofia muscular miotónica. Pueden comenzar a diferentes edades (generalmente durante la infancia) y causan debilidad y disminución del tono muscular. A menudo, se necesitan pruebas de diagnóstico (como una biopsia y pruebas genéticas) para diferenciar entre AME y distrofia muscular.
Botulismo: esta es una infección caracterizada por debilidad muscular severa, disminución del tono muscular y dificultad para respirar. El botulismo es causado por la exposición a las bacterias.Clostridium botulinum. Puede transmitirse a través de alimentos contaminados o heridas abiertas contaminadas. El botulismo puede afectar a personas de todas las edades y tiende a ser más grave en los niños que en los adultos (aunque los adultos también pueden tener efectos graves). Un examen físico puede diferenciar entre botulismo y AME.
Adrenoleucodistrofia: una enfermedad hereditaria rara, la adrenoleucodistrofia comienza durante la niñez y causa debilidad muscular y cambios en la visión, así como muchos problemas neurológicos. Esta enfermedad generalmente se caracteriza por un aumento del tono muscular en lugar del tono muscular disminuido típico de la AME. La adrenoleucodistrofia generalmente causa cambios reconocibles que se pueden ver en una resonancia magnética del cerebro.
Síndrome de Prader-Willi: esta afección hereditaria comienza en la primera infancia y puede causar debilidad muscular y disminución del tono muscular, así como efectos cognitivos y conductuales. Debido a que es causado por un defecto genético, se puede identificar con una prueba genética.
Síndrome de Angelman: una afección hereditaria que causa graves problemas de desarrollo, el síndrome de Angelman puede causar debilidad muscular en niños pequeños. Esta condición causa una gama más amplia de problemas neurológicos que la AME.
Miastenia gravis: esta es una afección autoinmune (el sistema inmunológico del cuerpo daña el propio cuerpo de una persona) que afecta la unión neuromuscular, que es el área entre un nervio y un músculo. Por lo general, causa caída de los párpados, pero puede causar debilidad de los músculos proximales y de los músculos respiratorios como la AME. La miastenia grave afecta a los adultos con más frecuencia que a los niños.
Neuropatía: hay una serie de neuropatías (enfermedades nerviosas) que afectan a los adultos con más frecuencia que a los niños. Las neuropatías causan debilidad muscular y disminución del tono muscular, y también pueden causar disminución de la sensibilidad.
Síndrome de Guillain-Barré (GBS): el síndrome de Guillain-Barré es una neuropatía progresiva que suele afectar a los adultos. Por lo general, causa debilidad en las piernas que puede extenderse rápidamente por el cuerpo y causar debilidad de los músculos respiratorios.
Esclerosis múltiple (EM): la EM generalmente afecta a adultos y no a niños. Puede causar una variedad de síntomas neurológicos, el más prominente de los cuales es la debilidad. La EM también suele tener efectos que no son característicos de la AME, como pérdida sensorial, pérdida de la visión y cambios cognitivos.
Esclerosis lateral amiotrófica (ELA): esta rara condición es, como la AME, una enfermedad de las neuronas motoras. Provoca debilidad muscular en adultos afectados. La ELA no afecta la visión, las sensaciones ni la cognición (pensamiento).
Puede ser difícil distinguir entre la AME de inicio en la edad adulta y la ELA. Las pruebas genéticas para el gen SMA pueden diferenciar entre las dos condiciones. La ELA tiene un peor pronóstico que la AME de inicio en la edad adulta.
Enfermedad de Kennedy: una enfermedad genética que a menudo se conoce como atrofia muscular espinobulbar (SBMA), la enfermedad de Kennedy es una enfermedad de la neurona motora que puede causar síntomas similares a los de la ELA y la AME de inicio en la edad adulta, incluida la debilidad de brazos y piernas. Esta afección se puede diagnosticar con una prueba genética.
Lo que debe saber sobre el tratamiento de la atrofia muscular espinal (AME)