Para la mayoría de los científicos, la vida tiene que ver con la reproducción. A nivel biológico, los organismos como los humanos, los hongos, las plantas y las bacterias pueden considerarse formas elaboradas a base de proteínas para que fragmentos de ácido desoxirribonucleico (ADN) se copien a sí mismos de manera más eficaz.
Paul Gilham / Personal / Getty ImagesDe hecho, el impulso de reproducirse incluso se extiende más allá de los organismos vivos. Los virus son un ejemplo de un extraño limbo entre los vivos y los no vivos. En cierto modo, un virus es poco más que una máquina reproductora.
En los casos de algunos virus, como el virus de la inmunodeficiencia humana (VIH), el ADN ni siquiera es la molécula que impulsa la reproducción. Otro nucleótido, el ARN (ácido ribonucleico), es el factor impulsor.
¿Qué es una enfermedad priónica?
Los priones (pronunciados pree-ons en los EE. UU., Pry-ons en el Reino Unido) están aún más alejados de los mecanismos de reproducción mejor entendidos que involucran al ADN y al ARN. El ADN y el ARN son nucleótidos, una estructura química utilizada para producir proteínas, los componentes básicos de los organismos vivos diseñados para garantizar una reproducción exitosa. Un prión es una proteína que no requiere un nucleótido para reproducirse; el prión es más que capaz de cuidarse a sí mismo.
Cuando una proteína prión plegada anormalmente se convierte en una proteína priónica normal, la proteína normal se transforma en otro prión causante de enfermedad plegado anormalmente. El resultado es una cascada incesante de proteínas mutadas. En los casos de enfermedad priónica hereditaria, es la mutación genética la que provoca un plegamiento anormal de la proteína priónica.
Desafortunadamente, estas son las mismas proteínas que utilizan las células cerebrales para funcionar correctamente, por lo que las células nerviosas mueren como resultado, lo que lleva a una demencia rápidamente progresiva. Si bien un prión que causa una enfermedad puede permanecer inactivo durante años, cuando los síntomas finalmente se hacen evidentes, la muerte puede sobrevenir en tan solo unos meses.
Hay cinco tipos principales de enfermedades priónicas actualmente reconocidas en humanos: enfermedad de Creutzfeldt-Jakob (CJD), enfermedad de Creutzfeldt-Jakob variante (vCJD), kuru, síndrome de Gerstmann-Straussler-Scheinker (GSS) e insomnio familiar fatal (FFI). Sin embargo, se están descubriendo formas más nuevas de enfermedad priónica.
Causas
Las enfermedades priónicas se pueden adquirir de tres formas: familiares, adquiridas o esporádicas. La forma más común de desarrollar una enfermedad priónica parece ser espontánea, sin una fuente de infección o herencia. Aproximadamente una de cada millón de personas desarrolla esta forma más común de enfermedad priónica.
Algunas enfermedades priónicas, como CJD, GSS y FFI, pueden heredarse. Otros se transmiten por contacto cercano con la proteína priónica. Por ejemplo, el kuru se difundió mediante rituales caníbales en Nueva Guinea. Cuando se comían cerebros como parte del ritual, se ingirieron los priones y la enfermedad se propagaría.
Un ejemplo menos exótico es la vCJD, que se sabe que se transmite de los animales a las personas cuando ingerimos la carne. Esto se conoce comúnmente como "enfermedad de las vacas locas" y ocurre cuando el prión existe en la vaca viva. También se ha descubierto que otros animales, como los alces y las ovejas, a veces albergan enfermedades causadas por priones. Si bien es poco común, las enfermedades priónicas también se pueden transmitir a través de instrumentos quirúrgicos.
Síntomas
Si bien todas las enfermedades causadas por priones causan síntomas ligeramente diferentes, todos los priones parecen tener una afición única por el sistema nervioso. Mientras que las infecciones bacterianas o virales se conocen comúnmente en muchas partes diferentes del cuerpo, incluido el cerebro, las enfermedades priónicas parecen causar exclusivamente síntomas neurológicos en humanos, aunque las proteínas mismas pueden encontrarse en una amplia gama de tejidos humanos. El tiempo puede mostrar que un mecanismo similar a un prión está detrás de las enfermedades fuera del cerebro.
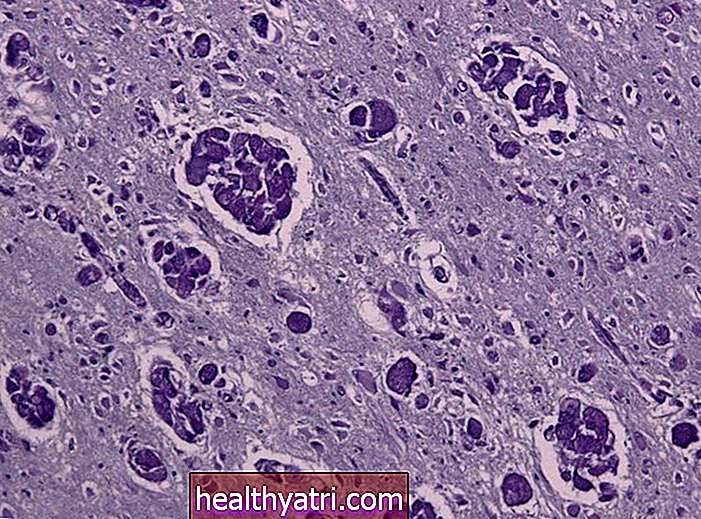
El impacto en el sistema nervioso es dramático. La mayoría de las enfermedades priónicas provocan lo que se conoce como encefalopatía espongiforme. La palabra espongiforme significa que la enfermedad erosiona el tejido cerebral, creando agujeros microscópicos que hacen que el tejido parezca una esponja.
Por lo general, el resultado final es una demencia rápidamente progresiva, lo que significa que la víctima pierde su capacidad de pensar como solía hacerlo en cuestión de meses a algunos años. Otros síntomas incluyen torpeza (ataxia), movimientos anormales como corea o temblor y patrones de sueño alterados.
Una de las cosas atemorizantes de la enfermedad priónica es que puede haber un período prolongado de incubación entre el momento en que alguien se expone a un prión y el momento en que desarrolla los síntomas. Las personas pueden pasar años antes de que los priones que han estado transportando se vuelvan obvios, con problemas neurológicos típicos.
Tratamiento
Desafortunadamente, no existe cura para la enfermedad priónica. En el mejor de los casos, los médicos pueden intentar ayudar a controlar los síntomas que causan malestar. En un pequeño estudio europeo, un medicamento provocó que la flupirtina (no disponible en los Estados Unidos) mejorara levemente el pensamiento en pacientes con ECJ, pero no mejoró su esperanza de vida. Un ensayo de los medicamentos clorpromazina y quinacrina no mostró mejoría. En este momento, las enfermedades causadas por priones siguen siendo universalmente fatales.