El rabdomiosarcoma es un cáncer de músculo esquelético poco común que se presenta con mayor frecuencia en la niñez. Es un tipo de sarcoma. Los sarcomas son tumores que surgen de las células mesoteliales, las células del cuerpo que dan lugar al tejido conectivo como huesos, cartílagos, músculos, ligamentos y otros tejidos blandos. En contraste, aproximadamente el 85% de los cánceres son carcinomas, que surgen de las células epiteliales.
A diferencia de los carcinomas en los que las células epiteliales tienen algo que se conoce como "membrana basal", los sarcomas no tienen una etapa de "células precancerosas" y, por lo tanto, las pruebas de detección de las etapas precancerosas de la enfermedad no son eficaces.
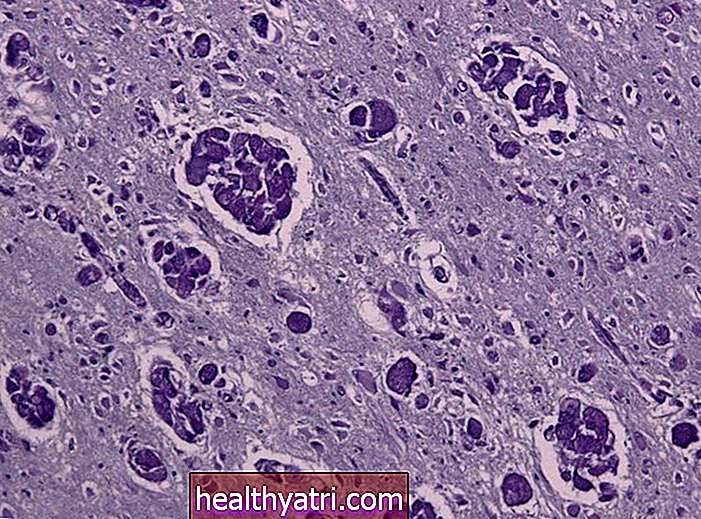
Frantab / Istockphoto.comEl rabdomiosarcoma es un cáncer de las células musculares, específicamente los músculos esqueléticos (músculos estriados) que ayudan en el movimiento de nuestro cuerpo. Históricamente, el rabdomiosarcoma se conoce como el “tumor de células pequeñas y redondas de color azul de la infancia” en función del color que adquieren las células con un tinte específico que se usa en los tejidos.
En conjunto, este cáncer es el tercer tipo más común de cáncer sólido infantil (sin incluir los cánceres relacionados con la sangre, como la leucemia). Es un poco más común en niños que en niñas y también es un poco más común en niños asiáticos y afroamericanos que en niños blancos.
Tipos
El rabdomiosarcoma se divide en tres subtipos:
- El rabdomiosarcoma embrionario representa del 60 al 70% de estos cánceres y ocurre con mayor frecuencia en niños entre las edades de nacimiento y los 4 años de edad. Este tipo se divide nuevamente en subtipos. Los tumores embrionarios pueden ocurrir en el área de la cabeza y el cuello, los tejidos genitourinarios u otras regiones del cuerpo.
- El rabdomiosarcoma alveolar es el segundo tipo más común y se encuentra en niños entre las edades de nacimiento y 19 años. Es el tipo más común de rabdomiosarcoma que se observa en adolescentes y adultos jóvenes. Estos cánceres a menudo se encuentran en las extremidades (brazos y piernas), el área genitourinaria, así como en el pecho, el abdomen y la pelvis.
- El rabdomiosarcoma anaplásico (pleomórfico) es menos común y se observa con más frecuencia en adultos que en niños.
Sitios
Los rabdomiosarcomas pueden ocurrir en cualquier parte del cuerpo donde esté presente el músculo esquelético. Las áreas más comunes incluyen la región de la cabeza y el cuello, como los tumores orbitarios (alrededor del ojo) y otras regiones, la pelvis (tumores genitourinarios), cerca de los nervios que excitan el cerebro (parameníngeos) y en las extremidades (brazos y piernas). ).
Signos y síntomas
Los síntomas del rabdomiosarcoma varían ampliamente según el sitio del tumor. Los síntomas por región del cuerpo pueden incluir:
- Tumores genitourinarios: los tumores en la pelvis pueden causar sangre en la orina o la vagina, una masa escrotal o vaginal, obstrucción y dificultades intestinales o vesicales.
- Tumores orbitarios: los tumores cerca del ojo pueden causar hinchazón alrededor del ojo y protuberancia del ojo (proptosis).
- Tumores parameníngeos: los tumores cerca de la médula espinal pueden presentarse con problemas relacionados con el nervio craneal al que están cerca, como dolor facial, síntomas de los senos nasales, hemorragia nasal y dolores de cabeza.
- Extremidades: cuando los rabdomiosarcomas se presentan en los brazos o las piernas, el síntoma más común es un bulto o hinchazón que no desaparece sino que aumenta de tamaño.
Incidencia
El rabdomiosarcoma es poco común y representa aproximadamente el 3,5% de los cánceres en niños. Alrededor de 250 niños son diagnosticados con este cáncer en los Estados Unidos cada año.
Causas y factores de riesgo
No sabemos exactamente qué causa el rabdomiosarcoma, pero se han identificado algunos factores de riesgo. Éstos incluyen:
- Niños con síndromes hereditarios como neurofibromatosis tipo 1 (NF1), síndrome de Li-Fraumeni, síndrome de Costello, blastoma pleuropulmonar, síndrome cutáneo cardio-facial, síndrome de Noonan y síndrome de Beckwith-Wiedermann
- Uso de marihuana o cocaína por parte de los padres
- Alto peso al nacer
Diagnóstico
El diagnóstico de rabdomiosarcoma generalmente comienza con una historia clínica y un examen físico cuidadosos. Dependiendo de la ubicación del tumor, se pueden realizar estudios de imágenes como radiografías, tomografías computarizadas, resonancias magnéticas, gammagrafías óseas o PET.
Para confirmar un diagnóstico sospechoso, generalmente es necesario realizar una biopsia. Las opciones pueden incluir una biopsia con aguja fina (usando una aguja pequeña para aspirar una muestra del tejido), una biopsia con aguja gruesa o una biopsia abierta. Una vez que el patólogo tiene una muestra del tejido, se observa el tumor bajo el microscopio y, a menudo, se realizan otros estudios para determinar el perfil molecular del tumor (buscando mutaciones genéticas responsables del crecimiento del tumor).
Para verificar si hay enfermedad metastásica, se puede realizar una biopsia de ganglio centinela. Esta prueba consiste en inyectar un tinte azul y un marcador radiactivo en el tumor y luego tomar una muestra de los ganglios linfáticos más cercanos al tumor que se iluminan o se tiñen de azul. También puede ser necesaria una disección completa de los ganglios linfáticos si los ganglios centinela dan positivo en la prueba de cáncer. Los estudios adicionales para buscar metástasis pueden incluir una gammagrafía ósea, una biopsia de médula ósea y / o una tomografía computarizada del tórax.
Puesta en escena y agrupación
La "gravedad" de un rabdomiosarcoma se define mejor al determinar la etapa o el grupo del cáncer.
Hay 4 etapas del rabdomiosarcoma:
- Estadio I: los tumores en estadio I se encuentran en "sitios favorables" como la órbita (alrededor del ojo), la cabeza y el cuello, en los órganos reproductores (como los testículos o los ovarios), los conductos que conectan los riñones con la vejiga (los uréteres ), el tubo que conecta la vejiga con el exterior (uretra) o alrededor de la vesícula biliar. Estos tumores pueden haberse diseminado o no a los ganglios linfáticos.
- Estadio II: los tumores en estadio II no se han diseminado a los ganglios linfáticos, no miden más de 5 cm (2 1/2 pulgadas), pero se encuentran en "sitios desfavorables", como cualquiera de los sitios no mencionados en el estadio I.
- Estadio III: el tumor estaba presente en un sitio desfavorable y puede o no haberse diseminado a los ganglios linfáticos o ser mayor de 5 cm.
- Estadio IV: cáncer de cualquier estadio o compromiso de los ganglios linfáticos que se ha diseminado a sitios distantes.
También hay 4 grupos de rabdomiosarcomas:
- Grupo I: el grupo 1 incluye tumores que se pueden extirpar por completo con cirugía y que no se han diseminado a los ganglios linfáticos.
- Grupo 2: los tumores del grupo 2 se pueden extirpar con cirugía, pero todavía hay células cancerosas presentes en los márgenes (en el borde del tumor extirpado) o el cáncer se ha diseminado a los ganglios linfáticos.
- Grupo 3: Los tumores del grupo 3 son aquellos que no se han diseminado, pero que no pueden extirparse con cirugía por algún motivo (a menudo debido a la ubicación del tumor).
- Grupo 4: Los tumores del grupo 4 incluyen aquellos que se han diseminado a regiones distantes del cuerpo.
Según las etapas y grupos anteriores, los rabdomiosarcomas se clasifican luego por riesgo en:
- Rabdomiosarcoma infantil de bajo riesgo
- Rabdomiosarcoma infantil de riesgo intermedio
- Rabdomiosarcoma infantil de alto riesgo
Metástasis
Cuando estos cánceres se diseminan, los sitios más comunes de metástasis son los pulmones, la médula ósea y los huesos.
Tratos
Las mejores opciones de tratamiento para el rabdomiosarcoma dependen del estadio de la enfermedad, el sitio de la enfermedad y muchos otros factores. Las opciones incluyen:
- Cirugía: la cirugía es el pilar del tratamiento y ofrece las mejores posibilidades de control a largo plazo del tumor. El tipo de cirugía dependerá de la ubicación del tumor.
- Radioterapia: la radioterapia se puede usar para encoger un tumor que no es operable o para tratar los bordes del tumor después de la cirugía para extirpar las células cancerosas restantes.
- Quimioterapia: los rabdomiosarcomas tienden a responder bien a la quimioterapia, y el 80% de estos tumores disminuye de tamaño con el tratamiento.
- Ensayos clínicos: otros métodos de tratamiento, como los medicamentos de inmunoterapia, se están estudiando actualmente en ensayos clínicos.
Albardilla
Dado que muchos de estos cánceres ocurren en la infancia, tanto los padres como el niño deben afrontar este diagnóstico inesperado y aterrador.
Para los niños mayores y los adolescentes que viven con cáncer, hay mucho más apoyo que en el pasado. Desde comunidades de apoyo en línea hasta retiros contra el cáncer diseñados específicamente para niños y adolescentes, hasta campamentos para el niño o la familia, hay muchas opciones disponibles. A diferencia del entorno escolar donde un niño puede sentirse único, y no en el buen sentido, estos grupos incluyen a otros niños y adolescentes o adultos jóvenes que de manera similar están lidiando con algo que ningún niño debería tener que enfrentar.
Para los padres, hay pocas cosas tan desafiantes como enfrentar el cáncer en su hijo. A muchos padres les encantaría nada menos que cambiar de lugar con sus hijos. Sin embargo, cuidar de uno mismo nunca es más importante.
Hay una serie de comunidades en persona y en línea (foros en línea y grupos de Facebook) diseñadas específicamente para padres de niños que enfrentan cáncer infantil e incluso rabdomiosarcoma en particular. Estos grupos de apoyo pueden ser un salvavidas cuando se da cuenta de que los amigos de la familia no tienen forma de entender por lo que está pasando. Conocer a otros padres de esta manera puede brindarle apoyo y, al mismo tiempo, brindarle un lugar en el que los padres pueden compartir los últimos avances en investigación. A veces es sorprendente cómo los padres a menudo conocen los últimos métodos de tratamiento incluso antes que muchos oncólogos comunitarios.
No minimice su papel como defensor. El campo de la oncología es amplio y crece día a día. Y nadie está tan motivado como el padre de un niño que vive con cáncer. Conozca algunas de las formas de investigar el cáncer en línea y tómese un momento para aprender a ser su propio defensor (o el de su hijo) de la atención del cáncer.
Pronóstico
El pronóstico de un rabdomiosarcoma varía mucho según factores como el tipo de tumor, la edad de la persona diagnosticada, la ubicación del tumor y los tratamientos recibidos. La tasa de supervivencia general a 5 años es del 70%, y los tumores de bajo riesgo tienen una tasa de supervivencia del 90%. En general, la tasa de supervivencia ha mejorado mucho en las últimas décadas.
Una palabra de Verywell
El rabdomiosarcoma es un cáncer infantil que se presenta en los músculos esqueléticos en cualquier lugar del cuerpo donde se encuentren estos músculos. Los síntomas varían según el sitio particular del tumor, así como los mejores métodos para diagnosticar el tumor. La cirugía es el pilar del tratamiento y, si el tumor se puede extirpar con cirugía, hay buenas perspectivas para el control a largo plazo de la enfermedad. Otras opciones de tratamiento incluyen radioterapia, quimioterapia e inmunoterapia.
Si bien la supervivencia del cáncer infantil está mejorando, sabemos que muchos niños sufren los efectos tardíos del tratamiento.El seguimiento a largo plazo con un médico familiarizado con los efectos secundarios a largo plazo del tratamiento es esencial para minimizar el impacto de estas afecciones.
Para los padres y los niños diagnosticados, participar en una comunidad de apoyo de otros niños y padres que enfrentan el rabdomiosarcoma no tiene precio, y en la era de Internet, ahora hay muchas opciones disponibles.